Hematopoiesis . Leukemia . Myeloproliferative Neoplasms . Kinase Inhibitors . Transgenic Mice . Familial Predisposition . Genomics
Experimental Hematology
Molecular pathogenesis of myeloproliferative neoplasms
Myeloproliferative neoplasms (MPN) are a group of blood diseases characterized by aberrant proliferation of precursors of the myeloid, erythroid and megakaryocytic lineages. They represent clonal stem cell disorders with a tendency towards leukemic transformation. Currently, no curative therapy is available. MPNs comprise3 entities: polycythemia vera (PV), essential thrombocythemia (ET) and primary myelofibrosis (PMF). The goal of our studies is to advance the understanding of the molecular events that initiate MPN and influence its progression to leukemia.A recurrent mutation Janus kinase 2 (JAK2) gene that substitutes a valine tophenylalanine at position 617 (JAK2-V617F) is present in a majority of patients withMPN, in particular PV. This mutation leads to activation of the Jak2 tyrosine kinaseand represents a driver for the proliferation of hematopoietic cells. Activating mutations calreticulin (CALR) and the thrombopoietin receptor (MPL) represent other frequent driver events. Despite this progress, several questions remain unsolved including how a single JAK2 mutation causes three different MPN phenotypes, what other genes might be involved and what determines the progression to acute leukemia. We are examining these questions by combining three approaches: molecular studies in patients with sporadic MPN, genetic analysis of familial MPN and transgenic mouse models that mimic the human disease.
Analysis of MPN disease initiation
The discovery of clonal hematopoiesis of indeterminate potential (CHIP) and the fact that JAK2-V617F is one of the most frequent CHIP mutations in healthy indi-viduals showed that acquisition of JAK2-V617F not the rate-limiting step in MPN pathogenesis, as only a fraction of individuals who carry JAK2-V617F eventually de-velop MPN disease. We are examining which factors promote the expansion of the JAK2-mutant clone to MPN disease initiation (Fig. 1). We determined that addition-al mutations in epigenetic regulator genes, e. g. Ezh2, or Dnmt3a, or inflammatory signals (e. g. IL-1β), as well as inherited predisposition mutations can favor clonal expansion and MPN disease initiation.
Familial predisposition for MPN
Familial syndromes resembling MPN can be grouped into two classes:
- Inherited disorders with high penetrance and polyclonal hematopoiesis (“class 1”).
- Hereditary predisposition to true MPN (“class 2”), with low penetrance, clonal hematopoiesis and occurrence of somatic mutations, e.g. in JAK2-V617F.
We identified for the first time a mutation in the erythropoietin (EPO) gene that causes familial polyclonal erythrocytosis with elevated erythropoietin levels. This mutation, a single-nucleotide deletion (c.32delG), introduces a frameshift in exon 2 that interrupts translation of the main EPO messenger RNA (mRNA) transcript but initiates excess production of erythropoietin from what is normally a noncoding EPO mRNA transcribed from an alternative promoter located in intron 1. Families with “class 2” phenotype are more common than generally assumed. These germ line mutations increase the likelihood of transition from JAK2-V617F CHIP to MPN disease. We are using genetic methods to identify such pre-disposing mutations.
Mouse models for MPN
We generated JAK2-V617F transgenic mice that express the human JAK2-V617F and develop MPN phenotype. A major focus of our research is to examine the na-ture of the MPN initiating stem cells and their interactions with the bone marrow microenvironment. We demonstrated that MPN can be initiated by transplanting single hematopoietic stem cells that carry JAK2-V617F as the sole genetic altera-tion and that loss of function mutations in Ezh2, Dnmt3a promoted, while loss of IL-1β, the inflammatory master regulator, decreased MPN disease initiation from single HSCs. We are using our mouse models for pre-clinical screening of Jak2 in-hibitors and other potential therapeutic agents. We also found that mutant JAK2 induces metabolic reprogramming of hematopoietic cells causing increased ener-gy requirement that can be targeted by inhibitors of metabolism (Rao et al 2019).
Connection to Clinical Practice
Prof. Jakob Passweg, Dr. Beatrice Drexler and Dr. Pontus Lundberg
Division of Hematoloy, University Hospital Basel
New therapeutic approaches to treat MPN: From bench to bedside
A collaborative study with Dr. S. Mendez-Ferrer showed that a β3-adrenergic receptor agonist re-stored Nestin-positive cells within the stem cell niche, and thereby normalized blood counts and improved myelofibrosis in our JAK2-V617F MPN mouse model (Arranz et al., Nature 512:78-81, 2014). Based on these observations, we performed a clin- ical phase-II study with the β-3 sympathomimetic agonist Mirabegron in 39 JAK2-V617F-positive MPN patients (Drexler et al., 2019). While the pri-mary end point (reduction of JAK2-V617F allele bur- den of ≥ 50 %) was not reached, we observed in-crease in Nestin-positive niche cells and decrease in reticulin fibrosis in Mirabegron-treated patients. These results are encouraging and show that Mi-rabegron can modify the microenvironment where the JAK2-mutant stem cells are maintained and thereby diminish the fibrotic manifestations of MPN.
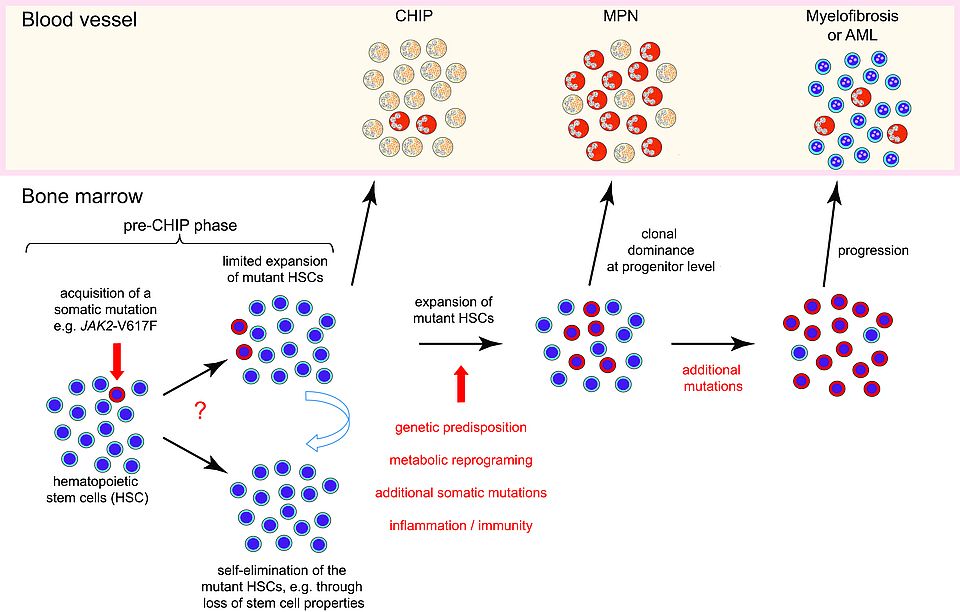
Fig. 1: Model of MPN disease initiation. Somatic mutations can occur in all cells including hematopoietic stem cells (HSCs). A single HSC in bone marrow that acquired a driver gene mutation (marked in red), e. g. JAK2-V617F, can either divide and increase the number of mutant HSCs, or divide and differentiate to mature cells of limited life span, thereby eliminating the mutant clone. This “pre-CHIP” phase is not yet detectable in peripheral blood. The JAK2-mutant clone has to expand to become self-sustaining and to generate sufficient numbers of mature blood cells to become detectable in peripheral blood (CHIP-phase). Further expansion and clonal dominance of the JAK2-mutant HSCs is required to initiate MPN disease characterized by increased numbers of erythrocytes and/or platelets (MPN-phase). Several factors (marked in red) can favor the transition from CHIP to MPN and the progression to myelofibrosis or acute leukemia (AML).